



Clues to future outbreaks may be hidden in existing genomic databases
Science
ELIZABETH PENNISI
26 JAN 2022
It took just one virus to cripple the world’s economy and kill millions of people; yet virologists estimate that trillions of still-unknown viruses exist, many of which might be lethal or have the potential to spark the next pandemic.
Now, they have a new-and very long-list of possible suspects to interrogate.
By sifting through unprecedented amounts of existing genomic data, scientists have uncovered more than 100,000 novel viruses, including nine coronaviruses and more than 300 related to the hepatitis Delta virus, which can cause liver failure.
“It’s a foundational piece of work,” says J. Rodney Brister, a bioinformatician at the National Center for Biotechnology Information’s National Library of Medicine who was not involved in the new study.
The work expands the number of known viruses that use RNA instead of DNA for their genes by an order of magnitude.
It also “demonstrates our outrageous lack of knowledge about this group of organisms,” says disease ecologist Peter Daszak, president of the EcoHealth Alliance, a nonprofit research group in New York City that is raising money to launch a global survey of viruses.
The work will also help launch so-called petabyte genomics-the analyses of previously unfathomable quantities of DNA and RNA data. (One petabyte is 10 15bytes.)
That wasn’t exactly what computational biologist Artem Babaian had in mind when he was in between jobs in early 2020.
Instead, he was simply curious about how many coronaviruses-aside from the virus that had just launched the COVID-19 pandemic-could be found in sequences in existing genomic databases.
So, he and independent supercomputing expert Jeff Taylor scoured cloud-based genomic data that had been deposited to a global sequence database and uploaded by the U.S. National Institutes of Health.
As of now, the database contains 16 petabytes of archived sequences, which come from genetic surveys of everything from fugu fish to farm soils to the insides of human guts. (A database with a digital photo of every person in the United States would take up about the same amount of space.) The genomes of viruses infecting different organisms in these samples are also captured by sequencing, but they usually go undetected.
To sift through the reams of data, Babaian and Taylor devised a set of computer tools specialized for searching cloud-based data.
With the help of several bioinformaticians, some whom became dedicated collaborators, they tweaked their software to make their analysis “way faster than anyone thought possible,” recalls Babaian, who is now at the University of Cambridge.
They soon expanded their viral hunt beyond coronaviruses and looked at all the data in the cloud. Babaian and colleagues performed their search by hunting for matches to the central core of the gene for RNA-dependent RNA polymerase, which is key to the replication of all RNA viruses. Such viruses include not only coronaviruses, but also those that cause flu, polio, measles, and hepatitis.
Babaian’s approach was fast enough to work through 1 million data sets a day-at a computing cost of less than 1 cent per data set.
“It’s an impressive engineering feat,” says C. Titus Brown, a bioinformatician at the University of California, Davis, who was not involved with the study.
When the researchers were finally finished, they had uncovered the partial genomes of almost 132,000 RNA viruses, they report today in Nature.
The group’s new database doesn’t have the complete sequence of each new virus-in many cases, there’s just the gene for the core enzyme.
But researchers can use even partial sequences to build family trees that reveal how different viruses are related and how they evolve.
They can also use the database to find out where a particular virus was found-and what its host is.
And some discoveries could help researchers better understand how human pathogens arise, Brown says, or improve diagnostic tests for viral infections.
Finally, when a new virus is isolated from a sick patient, researchers can more easily tell whether it has already been found elsewhere. “We have turned this [database] into a giant virus surveillance network,” Babaian says.
Some findings were unexpected, including previously unknown coronaviruses in the well-studied fugu fish and axolotls. In a few cases, researchers could piece together whole viral genomes. And in some aquatic animals, the sequences suggested the novel coronavirus genome has two separate loops, not the usual single RNA strand, Babaian and his colleagues report.
Babaian’s team also came across evidence of more than 250 giant viruses that infect bacteria and are similar to those found in algae.
Members of the bacteriophage viral group, close relatives of these “huge phages,” were detected in sequences from vastly different organisms.
One group of huge phages was found in a person in Bangladesh and also in cats and dogs in the United Kingdom, for example.
These viruses are big enough to carry genes between their host species, Babaian notes. That’s the way it is with viruses, Daszak says. “Every time we start digging, we get surprises.”
To make sure others can take advantage of the work, Babaian’s team has created a public repository of the tools it developed, along with the results.
The amount of cloud-based, publicly available DNA sequences is expanding exponentially; if he did the same analysis next year, Babaian says he would expect to find hundreds of thousands more RNA viruses.
“By the end of decade, I want to identify over 100 million.”
Names cited
J. Rodney Brister, a bioinformatician at the National Center for Biotechnology Information’s National Library of Medicine
disease ecologist Peter Daszak, president of the EcoHealth Alliance, a nonprofit research group in New York City
Computational biologist Artem Babaian
independent supercomputing expert Jeff Taylor scoured cloud-based genomic data
C. Titus Brown, a bioinformatician at the University of California, Davis
Originally published at https://www.science.org.
ORIGINAL PUBLICATION (long version)

Petabase-scale sequence alignment catalyses viral discovery
Nature
Robert C. Edgar, Jeff Taylor, Victor Lin, Tomer Altman, Pierre Barbera, Dmitry Meleshko, Dan Lohr, Gherman Novakovsky, Benjamin Buchfink, Basem Al-Shayeb, Jillian F. Banfield, Marcos de la Peña, Anton Korobeynikov, Rayan Chikhi & Artem Babaian
Abstract
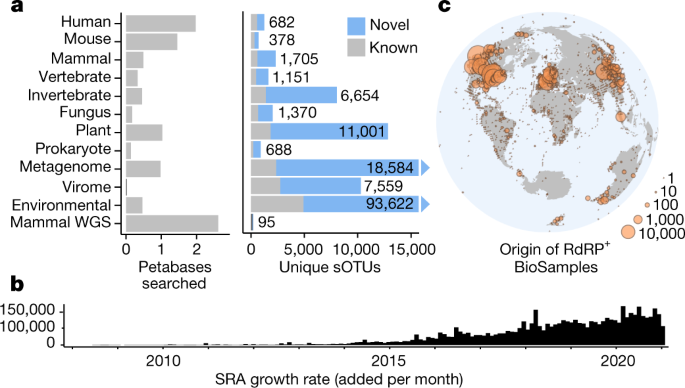
Public databases contain a planetary collection of nucleic acid sequences, but their systematic exploration has been inhibited by a lack of efficient methods for searching this corpus, which (at the time of writing) exceeds 20 petabases and is growing exponentially1.
Here we developed a cloud computing infrastructure, Serratus, to enable ultra-high-throughput sequence alignment at the petabase scale.
We searched 5.7 million biologically diverse samples (10.2 petabases) for the hallmark gene RNA-dependent RNA polymerase and identified well over 105 novel RNA viruses, thereby expanding the number of known species by roughly an order of magnitude.
We characterized novel viruses related to coronaviruses, hepatitis delta virus and huge phages, respectively, and analysed their environmental reservoirs.
To catalyse the ongoing revolution of viral discovery, we established a free and comprehensive database of these data and tools.
Expanding the known sequence diversity of viruses can reveal the evolutionary origins of emerging pathogens and improve pathogen surveillance for the anticipation and mitigation of future pandemics.
Fig. 1: Searching the planetary virome.
Originally published at https://www.nature.com







