




The Health Strategist
institute for continuous health transformation
and digital health
Joaquim Cardoso MSc
Chief Research and Strategy Officer (CRSO)
May 10, 2023
KEY MESSAGES
- The introduction of the pangenome as a more comprehensive genomic reference map marks a significant advancement in capturing the diversity and complexity of human genetic variation.
- It has the potential to enhance disease diagnosis, reveal previously hidden genetic insights, and promote more equitable representation in genomic research.
- However, further research and adoption of the pangenome approach are needed to fully realize its benefits and integrate it into existing genomic tools and practices.
ONE PAGE SUMMARY
This executive summary provides an overview of the latest development in genome mapping, known as the pangenome, which aims to capture all human genetic variation.
- The pangenome is a comprehensive genetic atlas that combines the complete DNA of 47 diverse individuals, representing various ethnic groups, into a single reference map.
- The project has been a decade-long effort, with plans to expand the dataset by incorporating DNA from an additional 300 individuals worldwide.
Traditional genome mapping efforts have relied on a single reference genome, which has limitations in representing the genetic diversity of the human population.
- The pangenome approach addresses this issue by utilizing a graph-based mathematical concept, where each DNA segment is represented as a dot connected through a network.
- This allows for a more accurate representation of individual genetic variations, including large differences such as deleted, inverted, or duplicated genes.
The pangenome has the potential to provide valuable insights into the diversity and uniqueness of individuals’ genomes.
- It could aid in the diagnosis of rare diseases and shed light on previously unexplored regions of the genome.
- However, the existing reference genome remains the primary tool for most biologists and medical professionals, as it is widely used and compatible with existing computational tools.
The publication of the pangenome is expected to address the issue of reference bias, which occurs when genetic information that deviates from the reference genome is overlooked during analysis.
- By incorporating more diverse genetic data, researchers hope to make gene research more equitable and ensure that individuals with genomes different from the current reference are adequately represented.
Constructing the pangenome involves the use of advanced sequencing technologies that read long stretches of DNA, resulting in more accurate assembly of genomes.
- Computational tools and algorithms have also been developed to organize and compare the vast amount of genomic data.
- While visualization tools like “spaghetti diagrams” have been used by specialists, the ultimate success of the pangenome lies in its seamless integration into genetic research infrastructure, without requiring researchers to directly interact with the complex underlying data representation.
It is important to note that the pangenome will always remain an ongoing and evolving project, as every individual and population may have their own unique genomic variations.
- While experts anticipate challenges in transitioning from a linear representation of the genome to a pangenome model, they believe it holds immense potential for advancing our understanding of human genetics.
DEEP DIVE

This new genome map tries to capture all human genetic variation
It’s called a pangenome, and it could explain the DNA that makes each of us unique.
MIT Technology Review
By Antonio Regalado
May 10, 2023
The joke about the Human Genome Project is how many times it’s been finished, but not actually.
The first time was in 2000, when Bill Clinton announced the “first survey of the entire human genome” at a White House ceremony, calling it “the most important and most wondrous map ever produced by humankind.”
But the job wasn’t done. A year later, the triumph was announced again, this time with the formal publication of a “draft” of “the genetic blueprint for a human being.” In 2003, researchers had another go at the finish line, claiming the “successful completion” of the project, citing better levels of accuracy. Nineteen years later, in 2022, they again claimed victory, this time for a really, truly “complete” sequence of one genome-end to end, no gaps at all. Pinkie promise.
Today, researchers announced yet another version of the human genome map, which they say combines the complete DNA of 47 diverse individuals-Africans, Native Americans, and Asians, among other groups-into one giant genetic atlas that they say better captures the surprising genetic diversity of our species.
The new map, called a “pangenome,” has been a decade in the making, and researchers say it will only get bigger, creating an expanding view of the genome as they add DNA from another 300 people from around the globe. It was published in the journal Nature today.
“We now understand that having one map of a single human genome cannot adequately represent all of humanity,” says Karen Miga, a professor at the University of California, Santa Cruz, and a participant in the new project.
Diversity in detail
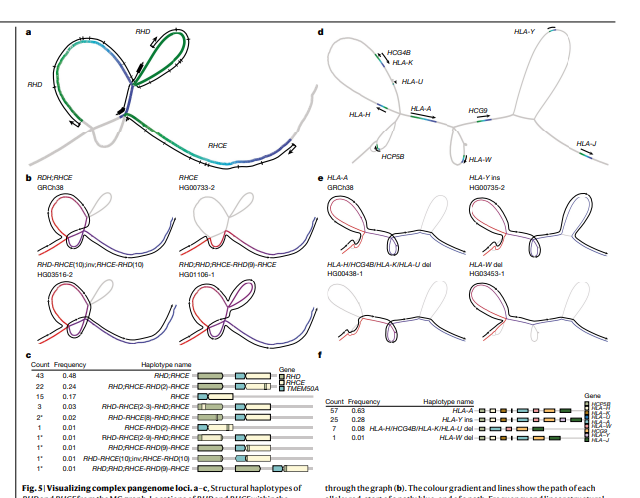
People’s genomes are largely alike, but it’s the hundreds of thousands of differences, often just single DNA letters, that explain why each of us is unique. The new pangenome, researchers say, should make it possible to observe this diversity in more detail than ever before, highlighting so-called evolutionary hot spots as well as thousands of surprisingly large differences, like deleted, inverted, or duplicated genes, that aren’t observable in conventional studies.
The pangenome relies on a mathematical concept called a graph, which you can imagine as a massive version of connect-the-dots. Each dot is a segment of DNA. To draw a particular person’s genome, you start connecting the numbered dots. Each person’s DNA can take a slightly different path, skipping some numbers and adding others.
One payoff of the new pangenome could be better ways to diagnose rare diseases, although practical applications aren’t easy to name. Instead, scientists say it’s mainly giving them insight into some of the “dark matter” of the genome that’s previously been hard to see, including strange regions of chromosomes that seem to share and exchange genes.
For now, most biologists and doctors will stick to the existing “reference genome,” the one first produced in draft form in 2001 and gradually improved. It answers most questions researchers are interested in, and all their computer tools work with it.
The reason a reference genome is important is that when a new person’s genome is sequenced, that sequence is projected onto the reference in order to organize and read the new data. Yet since the current reference is just one possible genome, missing bits that some people have, some information can’t be analyzed and is usually ignored.
Researchers call this effect “reference bias” or, more simply, the streetlamp problem. You don’t see where you don’t look.
“It’s hard to appreciate just how important the current reference is. We use it like a coordinate system or a map, and we refer to it constantly when we talk about genes,” says Benedict Paten, a computational biologist, also at Santa Cruz, and the senior author of the report.. “But it’s both incomplete and lacks diversity. It lacks the things that make us different-in other words, the interesting bits.”
Officials with NIH said they hoped the new update to the genome map would make gene research more “equitable.” That’s because the more different your genome is from the current reference, the more information about you could be missed. The existing reference is largely the DNA of one African-American man, although it includes segments from several other people as well.
“If the genome you want to analyze has sequences that are not in that reference, they will be missed in the analysis,” says Deanna Church, a consultant with the business incubator General Inception, who previously held a key role at NIH managing the reference genome. “In reality, the notion that there is a ‘human genome’ is really the problem,” she says. “The current version is the simplest model you can make. It made sense when we started … But now we need better models.”
Piecing together the puzzle of us
The pangenome, which itself remains at draft stage, was constructed with the help of two newer technologies. One is a type of sequencing machine that reads out very long stretches of DNA in one go. Most sequencing is done by shredding DNA into tiny bits, under 200 letters long. But the new machines, made by the company Pacific Biosciences, produce continuous readouts of 10,000 letters at once.
Such “long reads,” as researchers call them, are like extra-large puzzle pieces that are much easier to arrange correctly in the actual order they’re present in a person’s genome.
That puzzling-together process-called genome assembly-is the other area where researchers say they’ve made advances with new computation tools. Even so, organizing and comparing 47 genomes at once (each with about 6 billion pairs of DNA letters) remains a gnarly problem.
“There is a huge amount of really interesting computer science that has been published in not-so-glamorous journals,” says Paten, who has been working on the pangenome for more than 10 years.
Paten also admits that no one other than specialists will want to look at their data visualization tools, which display the alternate arrangements of DNA as complicated loops and knots called “spaghetti diagrams.” Instead, real success will come if the pangenome can fade into the background and become the new plumbing of the genetic age, something researchers can use without ever seeing.
Experts think it’s too soon to say whether that will happen. “I hope it will, but it will be a tough road,” Church says. “So much of our tooling and infrastructure is based on having a linear representation that getting people to change their mindset will be hard.”
One thing is for sure, says Erik Garrison, a computational biologist at the University of Tennessee and also among the leaders of the project. The human genome isn’t finished and never will be.
“Once you start talking about a pangenome, it’s always going to be incomplete, and it’s never going to end. Every individual is going to have a different genome, so it’s an infinite process,” says Garrison. “Every population and every generation could have its own pangenome.”
Originally published at
ORIGINAL PUBLICATION

A draft human pangenome reference
Wen-Wei Liao, Mobin Asri, Jana Ebler, Daniel Doerr, Marina Haukness, Glenn Hickey, Shuangjia Lu, Julian K. Lucas, Jean Monlong, Haley J. Abel, Silvia Buonaiuto, Xian H. Chang, Haoyu Cheng, Justin Chu, Vincenza Colonna, Jordan M. Eizenga, Xiaowen Feng, Christian Fischer, Robert S. Fulton, Shilpa Garg, Cristian Groza, Andrea Guarracino, William T. Harvey, Simon Heumos, …Benedict Paten — Show authors
Abstract
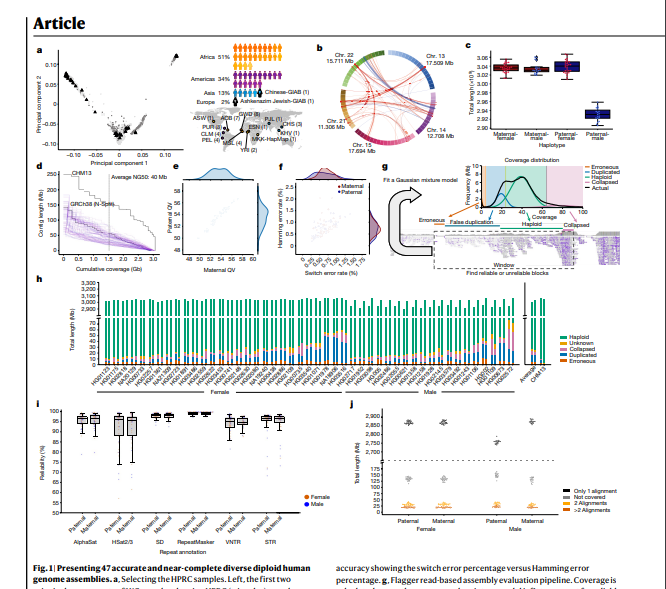
- Here the Human Pangenome Reference Consortium presents a first draft of the human pangenome reference.
- The pangenome contains 47 phased, diploid assemblies from a cohort of genetically diverse individuals1.
- These assemblies cover more than 99% of the expected sequence in each genome and are more than 99% accurate at the structural and base pair levels.
- Based on alignments of the assemblies, we generate a draft pangenome that captures known variants and haplotypes and reveals new alleles at structurally complex loci.
- We also add 119 million base pairs of euchromatic polymorphic sequences and 1,115 gene duplications relative to the existing reference GRCh38.
- Roughly 90 million of the additional base pairs are derived from structural variation.
- Using our draft pangenome to analyse short-read data reduced small variant discovery errors by 34% and increased the number of structural variants detected per haplotype by 104% compared with GRCh38-based workflows, which enabled the typing of the vast majority of structural variant alleles per sample.
Selected image(s)
Originally published at https://www.nature.com/







