




This is a republication of the paper “The next generation of evidence-based medicine — Deep Medicine, AI enabled”, with the title above, highlithing the topic in question.

The next generation of evidence-based medicine — Deep Medicine, AI enabled
“Every major disease needs a ‘Moonshot’ program and every rare disease should have an ‘Operation Warp Speed”
This is a republication of the paper “The next generation of evidence-based medicine”, with the title above. The title is preceded by an Executive Summary.
health transformation institute
clinical trials unit
Joaquim Cardoso MSc
Senior Advisor and
Chief Researcher & Editor
January 16, 2023
EXECUTIVE SUMMARY
Abstract
Recently, advances in wearable technologies, data science and machine learning have begun to transform evidence-based medicine, offering a tantalizing glimpse into a future of next-generation ‘deep’ medicine.
- Despite stunning advances in basic science and technology, clinical translations in major areas of medicine are lagging.
- While the COVID-19 pandemic exposed inherent systemic limitations of the clinical trial landscape, it also spurred some positive changes, including new trial designs and a shift toward a more patient-centric and intuitive evidence-generation system.
- In this Perspective, the authors shares his heuristic vision of the future of clinical trials and evidence-based medicine.
Conclusion
The success of future clinical trials requires a fundamental transformation in how trials are designed, conducted, monitored, adapted, reported and regulated to generate the best evidence.
- The status quo model is unsustainable.
- Instead, preventive, personalized, pragmatic and patient-participatory medicine is needed, and paradigm shifts are required to get there via sustainable growth.
- Silos need to be broken.
- Standards of care and clinical trials are currently viewed in different realms; however, the overarching goal of both is to improve health outcomes.
The COVID-19 pandemic created an opportunity to observe how routine clinical care and clinical trials can work synergistically to generate evidence86.
- Pragmatic platform trials such as the RECOVERY trial should be a model and guide for trial efficiency and real-time impact.
Current paradigms must be continuously challenged by emerging technology and by all stakeholders (the new generations of scientists, physicians, the pharma industry, regulatory authorities and, most importantly, patients).
- Disruptive innovation should lead to every clinical site being a research site, with all necessary quality checks and research as part of the standard of care.
- The healthcare system should be integrated into an intuitive RWE-generation system, with clinical research and clinical care going hand in hand.
- Beyond an ad hoc creative flash of genius (necessitated by a pandemic), sustained momentum will be needed to leverage the knowledge gained from programs such as ‘Operation Warp Speed’ (initiated by the US government to accelerate COVID-19 vaccine development).
- The personal view of the author is that every major disease needs a ‘Moonshot’ program and every rare disease should have an ‘Operation Warp Speed’-both with clearly identified, sustainable goals to improve population health and address equity, diversity and global access to therapies.
- Methodological advances and future AI-based analyses of all data will provide deep evidence to realize the goal of personalized medicine- that is, to offer the right treatment to the right patient at the right time.
The personal view of the author is that every major disease needs a ‘Moonshot’ program and every rare disease should have an ‘Operation Warp Speed’-both with clearly identified, sustainable goals to improve population health and address equity, diversity and global access to therapies.
INFOGRAPHIC
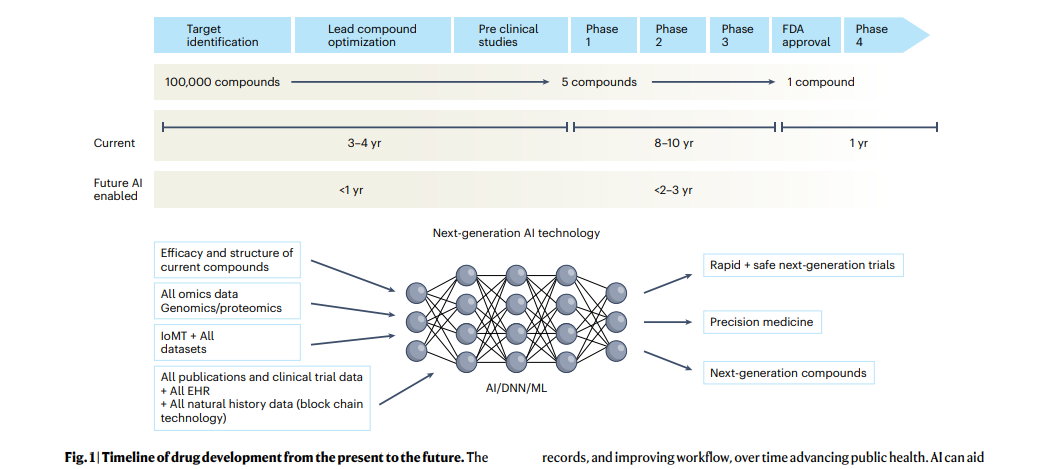
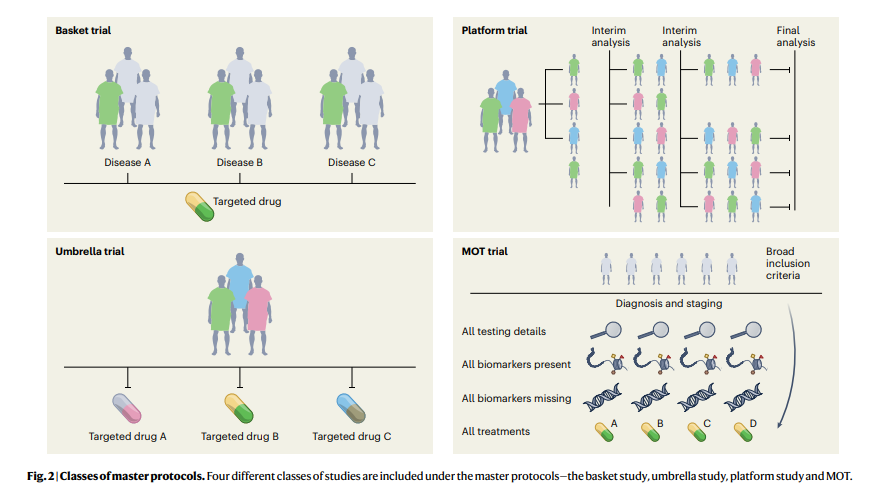
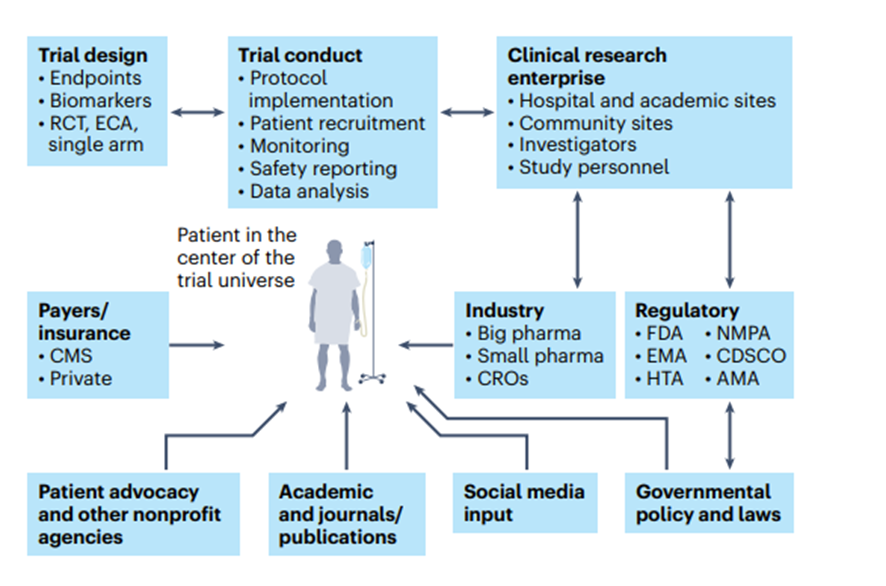
Fig. 3 | The patient as the center of the clinical trial universe in the clinical research enterprise.
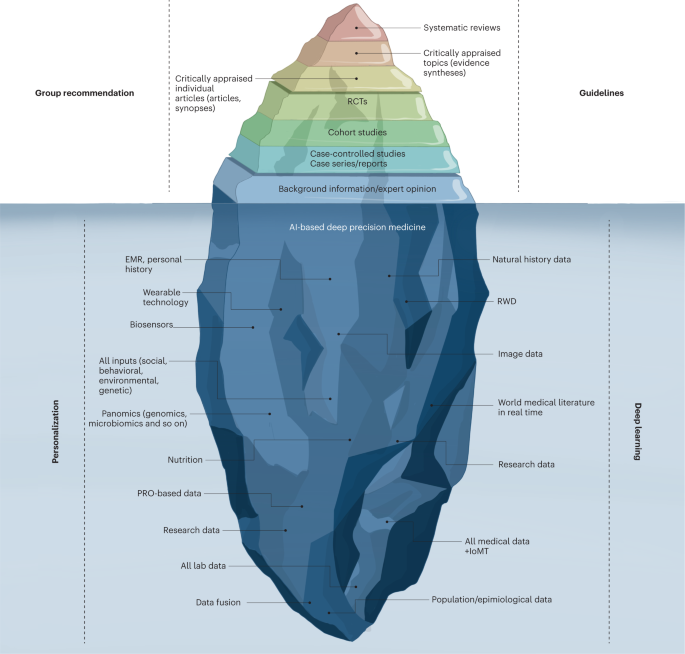
Fig. 4: Evidence-based deep medicine iceberg.
DEEP DIVE

Nature Medicine
Vivek Subbiah
16 January 2023
Main
The last 30 years have witnessed breathtaking, unparalleled advancements in scientific research-from a better understanding of the pathophysiology of basic disease processes and unraveling the cellular machinery at atomic resolution to developing therapies that alter the course and outcome of diseases in all areas of medicine.
Moreover, exponential gains in genomics, immunology, proteomics, metabolomics, gut microbiomes, epigenetics and virology in parallel with big data science, computational biology and artificial intelligence (AI) have propelled these advances. In addition, the dawn of CRISPR-Cas9 technologies has opened a tantalizing array of opportunities in personalized medicine.
Despite these advances, their rapid translation from bench to bedside is lagging in most areas of medicine and clinical research remains outpaced.
The drug development and clinical trial landscape continues to be expensive for all stakeholders, with a very high failure rate. In particular, the attrition rate for early-stage developmental therapeutics is quite high, as more than two-thirds of compounds succumb in the ‘valley of death’ between bench and bedside1,2. To bring a drug successfully through all phases of drug development into the clinic costs more than 1.5–2.5 billion dollars (refs. 3, 4). This, combined with the inherent inefficiencies and deficiencies that plague the healthcare system, is leading to a crisis in clinical research. Therefore, innovative strategies are needed to engage patients and generate the necessary evidence to propel new advances into the clinic, so that they may improve public health. To achieve this, traditional clinical research models should make way for avant-garde ideas and trial designs.
Before the COVID-19 pandemic, the conduct of clinical research had remained almost unchanged for 30 years and some of the trial conduct norms and rules, although archaic, were unquestioned.
The pandemic exposed many of the inherent systemic limitations in the conduct of trials5 and forced the clinical trial research enterprise to reevaluate all processes — it has therefore disrupted, catalyzed and accelerated innovation in this domain6,7. The lessons learned should help researchers to design and implement next-generation ‘patient-centric’ clinical trials.
Chronic diseases continue to impact millions of lives and cause major financial strain to society8, but research is hampered by the fact that most of the data reside in data silos.
The subspecialization of the clinical profession has led to silos within and among specialties; every major disease area seems to work completely independently. However, the best clinical care is provided in a multidisciplinary manner with all relevant information available and accessible. Better clinical research should harness the knowledge gained from each of the specialties to achieve a collaborative model enabling multidisciplinary, high-quality care and continued innovation in medicine. Because many disciplines in medicine view the same diseases differently-for example, infectious disease specialists view COVID-19 as a viral disease while cardiology experts view it as an inflammatory one-cross-discipline approaches will need to respect the approaches of other disciplines. Although a single model may not be appropriate for all diseases, cross-disciplinary collaboration will make the system more efficient to generate the best evidence.
Over the next decade, the application of machine learning, deep neural networks and multimodal biomedical AI is poised to reinvigorate clinical research from all angles, …
… including drug discovery, image interpretation, streamlining electronic health records, improving workflow and, over time, advancing public health (Fig. 1).
In addition, innovations in wearables, sensor technology and Internet of Medical Things (IoMT) architectures offer many opportunities (and challenges) to acquire data9.
In this Perspective, I share my heuristic vision of the future of clinical trials and evidence generation and deliberate on the main areas that need improvement in the domains of clinical trial design, clinical trial conduct and evidence generation.
The figure represents the timeline from drug discovery to first-in-human phase 1 trials and ultimately FDA approval.
- Phase 4 studies occur after FDA approval and can go on for several years.
- There is an urgent need to reinvigorate clinical trials through drug discovery, interpreting imaging, streamlining electronic health records, and improving workflow, over time advancing public health.
- AI can aid in many of these aspects in all stages of drug development.
- DNN, deep neural network; EHR, electronic health records; IoMT, internet of medical things; ML, machine learning.
Clinical trial design
Trial design is one of the most important steps in clinical research-better protocol designs lead to better clinical trial conduct and faster ‘go/no-go’ decisions. Moreover, losses from poorly designed, failed trials are not only financial but also societal.
Challenges with randomized controlled trials
Randomized controlled trials (RCTs) have been the gold standard for evidence generation across all areas of medicine, as they allow unbiased estimates of treatment effect without confounders.
Ideally, every medical treatment or intervention should be tested via a well-powered and well-controlled RCT. However, conducting RCTs is not always feasible owing to challenges in generating evidence in a timely manner, cost, design on narrow populations precluding generalizability, ethical barriers and the time taken to conduct these trials. By the time they are completed and published, RCTs become quickly outdated and, in some cases, irrelevant to the current context. In the field of cardiology alone, 30,000 RCTs have not been completed owing to recruitment challenges10. Moreover, trials are being designed in isolation and within silos, with many clinical questions remaining unanswered. Thus, traditional trial design paradigms must adapt to contemporary rapid advances in genomics, immunology and precision medicine11.
Progress in clinical trial design
High-quality evidence is needed for clinical practice, which has traditionally been achieved with RCTs12.
In the last decade, substantial progress has been made in the design, conduct and implementation of ‘master’ protocols (overarching protocols that apply to several substudies), which has led to many practice changes that have substantially improved the stagnation of RCTs. Moreover, master protocols may involve parallel interventional studies in a single disease or multiple diseases defined by a biomarker or disease entity12. Four different classes of studies are included under the master protocols-the umbrella study, basket study, platform study and master observational trial (MOT) (Fig. 2). Each of these is a unique trial design that can include independent arms with control interventions and may be analyzed individually and/or collectively, with added flexibility13,14. The field of oncology has led these efforts more so than any other field, owing to advances in genomics (for identifying molecular alterations), discovery of therapeutics and rapid clinical translation, thus ushering in the precision oncology era.
Four different classes of studies are included under the master protocols — the basket study, umbrella study, platform study and MOT.
1.Umbrella study
Umbrella trials are study designs that evaluate multiple targeted therapies for the same disease entity, stratified by molecular alteration.
Examples include the I-SPY (Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging And Molecular Analysis) breast cancer trial and Lung-MAP (Lung Cancer Master Protocol)15,16.
2.Basket (or bucket) trial
Basket trials are tissue-agnostic or histology-independent studies where targeted therapy is evaluated on multiple disease types that all harbor the same underlying molecular aberration.
For instance, the VE-Basket study (in which VE denotes vemurafenib)17, Rare Oncology Agnostic Research (ROAR) study18, ARROW trial19and LIBRETTO-001 trials20,21have led to several drug approvals in specific biomarker-driven populations in a histology-dependent and histology-independent manner.
3.Platform study
These are multi-arm, multistage study designs that compare several intervention groups with a common control group in the context of the same master protocol.
Additionally, they can be perpetual/immortal study designs (with no defined end date) and are more efficient than traditional trials on account of the shared control arm, which ensures that a greater proportion of patients are enrolled in the interventional/experimental arms than in the control arm. The Randomised Evaluation of COVID-19 Therapy (RECOVERY) Platform Study is a prominent example; this practice-changing trial established dexamethasone as an effective treatment for COVID-19 (ref. 22) and also showed that hydroxychloroquine was ineffective. Platform studies are flexible by design and do not necessarily need to have a shared control arm; the main idea is that intervention arms may be added to an ongoing trial, for example, as in the The UK Plasma Based Molecular Profiling of Advanced Breast Cancer to Inform Therapeutic CHoices (plasmaMATCH) platform trial23. Although the aforementioned trials were designed in the context of drug development in oncology and infectious diseases, the scope of platform trials could be leveraged in other diverse areas such as clinical psychology and neurology24. Such trials could also be used for digital mental health interventions and could be readily implemented in resource-constrained settings24.
4.MOT
The MOT is a prospective, observational study design that broadly accepts patients independently of biomarker signature and collects comprehensive data on each participant14,25.
The MOT is a combination of the master interventional trial and prospective observational trial designs and attempts to hybridize the power of biomarker-based master interventional protocols with the breadth of real-world data (RWD)14,25. This approach could be well suited to collect prospective RWD across many specialties; the Registry of Oncology Outcomes Associated with Testing and Treatment (ROOT) MOT is one example14.

Development of biomarkers and defining endpoints
Biomarker development has facilitated progress in clinical trial design, with unprecedented advances in genomics and immunology leading to several approvals for biomarker-based targeted therapies and immunotherapy in the last decade.
In fact, human genetics evidence provided support for more than two-thirds of the drug approvals in 2021 (ref. 26).
The fields of oncology and genetics have benefited immensely from these advances, but fields such as cardiology, nephrology and pulmonology are still lagging in biomarker-based drug approvals.
To fast-track drug development and clinical trials in every major disease, we will need to define biomarkers (whether clinical, pathological or physiological) and their context of use for every disease process and delineate clear endpoints for studies27.
Biomarkers can be diagnostic, prognostic or predictive and can inform early drug development, dose selection and trial design.
In addition, biomarkers can help to fast-track basic science and drug discovery — all with the eventual goal of improving patient health28.
However, the level of evidence for a biomarker largely depends on the context of use.
In addition to biomarkers, every field needs to define areas of top priority for research and identify the most relevant endpoints to answer priority research questions.
Endpoints are measures of health and/or disease and serve different purposes depending on the phase of the trial28,29.
Beyond clinical and regulatory endpoints, patient-reported outcomes and digital endpoints are also rapidly emerging.
Digital endpoints
Digital endpoints are sensor-generated data collected outside the clinical environment in the context of patients’ routine living-such as using smartphone microphones to monitor cognitive decline in people with Alzheimer’s disease or smartwatch monitors to evaluate drug effect in people with sickle-cell anemia29.
This is an area of considerable excitement in medicine as it could permit more realistic real-world tracking of the patient experience.
Moreover, with the increase in decentralized trial conduct across many specialties, remote monitoring is poised to increase.
For instance, a recent study developed an AI model to detect and track progression of Parkinson’s disease (for which there are no biomarkers) on the basis of nocturnal breathing signals using noninvasive, at-home assessment, providing evidence that AI may be useful in risk assessment before clinical diagnosis of the condition29,30.
Additionally, digital atrial fibrillation screening by smart devices has been evaluated extensively in large-scale studies, including the Apple31, Huawei32and Fitbit33cardiac studies.
Altogether, these siteless observational studies enrolled over 1 million participants, an amazing feat, and a randomized study showed the superiority of digital atrial fibrillation detection over usual care34.
Digital characterization and assessment of clinical status need to be standardized and harmonized, with interdisciplinary collaboration and regulatory input.
Consensus is also needed to identify and characterize intermediate and surrogate endpoints for major chronic diseases.
This requires specialty-specific incorporation of multiple levels of data such as genomic, proteomic and genotype–phenotype-based clinical data and disease-specific measurements, in addition to a layer of functional data26.
The National Institutes of Health (NIH) and Food and Drug Administration (FDA) have developed BEST (Biomarkers, EndpointS and other Tools) resources to clarify the ambiguity in biomarkers and endpoints.
This is a ‘living document’ that is continually updated as standards and evidence change35 and that clarifies important definitions and describes some of the hierarchical relationships, connections and dependencies among the terms.
Clinical trial conduct
The components of clinical trial conduct are protocol implementation; patient selection, recruitment, monitoring and retention; ensuring compliance to safety reporting; and continuing review and data analysis.
The pharmaceutical industry and the healthcare sector invest substantial resources into clinical trial conduct, but changes are urgently needed to make the process more seamless.
Moreover, the pace at which clinical trials are conducted is too slow to match the research advances in every field; thus, a high-tech transformation of every component in a stepwise manner is needed.
One of the positive sides of the pandemic is that it forced the system to redirect clinical trials to be more patient-centric than before, thus giving more importance to the principal subject of clinical research-the patient36(Fig. 3).
This has led to decentralized trials and digital, remote and ‘virtual’ trials (which allow patients access to trials regardless of their geographic location), as well as ‘hospital-at-home’ and home-based monitoring concepts37.
Such rapid strides have been aided by guidance from regulatory authorities38.
Adopting an AI-based approach to enhance the patient experience can further improve high-fidelity assessments and ensure compliance with protocols39.
Although digitalization, virtualization and decentralization are not cures for clinical research crises, they can create efficiencies that may have a sizeable and long-term downstream impact.
Adopting an AI-based approach to enhance the patient experience can further improve high-fidelity assessments and ensure compliance with protocols39.
Although digitalization, virtualization and decentralization are not cures for clinical research crises, they can create efficiencies that may have a sizeable and long-term downstream impact.
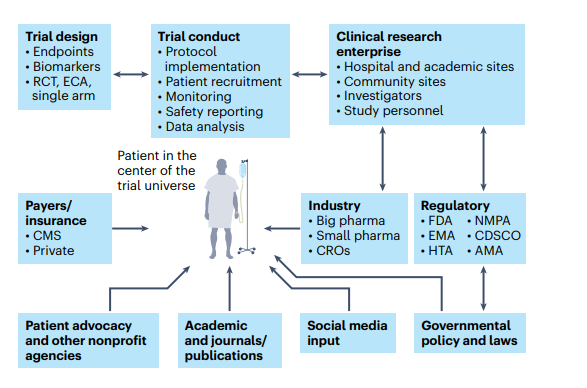
Fig. 3 | The patient as the center of the clinical trial universe in the clinical research enterprise.
The main constituents of the clinical trial enterprise — patients, academic centers, industry sponsors (big and small pharma), government/cooperative group sponsors, regulatory agencies, patient advocacy organizations and CROs — need to work together, with the patient as the center of this clinical trial universe.
AMA, African Medicines Agency; CDSCO, Central Drugs Standard Control Organization (India); CMS, Centers for Medicare and Medicaid Services; ECA, external control arm; EMA, European Medicines Agency; HTA, Health Technology Assessment; NMPA, National Medical Products Administration (China).
Physicians, healthcare team members and clinical investigators at academic sites and other trial enrolling sites contribute immensely to patient recruitment.
In addition, high-impact, high-functioning sites (as in major academic centers of excellence) often have a portfolio of trials and screen patients presenting to the system in an efficient manner.
Such sites are in the minority, however, and most clinical trial sites are challenged with staffing constraints and other barriers.
Clinical trial research enterprise
Efficiency and collaboration in the clinical trial research enterprise are major components of clinical trial success.
The main constituents of the clinical trial enterprise are patients, academic centers, industry sponsors (big and small pharma), government/cooperative group sponsors, regulatory agencies, patient advocacy organizations and contract research organizations (CROs), and all of these need to work together with the patient as the center of the clinical trial universe (Fig. 3).
Moreover, this whole system needs a digital overhaul as many sites still use protocol binders, pen-and-paper diaries, faxes between sites, unstructured data and decades-old software systems.
Registrational clinical trials need to be well managed on a day-to-day basis with rigorous electronic data capture and monitoring.
Integration of blockchain technology into the clinical trial management system could conceivably bolster trust in the clinical trial process and facilitate regulatory oversight40.
Patient participation in clinical trials is key, as there can be no trials without patients.
Clinical trial organizers should make it easier for patients to participate in trials.
In addition, physician-patient treatment decisions for major diseases should include clinical trial options as standard.
These clinical trials should be easily accessible and should ensure that no patients are unnecessarily excluded; this can be achieved with site-agnostic clinical trial matching and navigation services.
In addition, clinical trial training should be a part of medical education so that a diverse pool of trained investigators and personnel from the entire healthcare enterprise can be available for clinical research.
It is about time
Clinical development timelines for drug candidates are a race against time from when patents are filed to final FDA approval41.
Drug development timelines, on average, are approximately 10 years (Fig. 1).
The swiftness of the development of the COVID-19 mRNA vaccines and the oral COVID-19 treatment nirmatrelvir/ritonavir tablets, both of which were developed within a year using a ‘lightspeed approach’, should not be an outlier42.
The lessons learned should provide a model for multiple therapeutic areas of unmet need.
The two small molecules that hold the record for the shortest timeline in drug development, osimertinib for EGFR-mutant non-small-cell lung cancer (NSCLC) (984 days via accelerated approval) and elexacaftor for cystic fibrosis (1,043 days via the regular path)41, in nonpandemic times demonstrate that this is possible.
The swiftness of the development of the COVID-19 mRNA vaccines and the oral COVID-19 treatment nirmatrelvir/ritonavir tablets, both of which were developed within a year using a ‘lightspeed approach’, should not be an outlier42.
The regulatory logjams slowing drug development necessitated the creation of programs such as the FDA’s accelerated approval pathway, which was introduced in 1992 to address the HIV and AIDS crisis and has since benefitted highly specialized areas such as precision oncology43.
Multiple programs have been created to shorten timelines for the premarket process, including priority review, fast-track designation, breakthrough designation and orphan designation44.
Beyond these programs, however, the timelines are still slow and there is an urgent need to address this for all diseases as drug development speed is crucial for patients, physicians and drug development stakeholders alike.
Globalizing drug development, harmonization and transportability
Although the mandate of the FDA is to the US population, their influence is global and, functionally, the FDA is the de facto regulator for the world.
Other regulatory authorities such as the European Medicines Agency, the National Medical Products Administration in China and the Central Drugs Standard Control Organization in India, which in total serve more than 3 billion of the world’s population, are also evolving as key players in the global pharmaceutical sector.
In addition, the newly established African Medicines Agency was set up (in 2019) to speed up timelines for vaccines and medicine approvals and to improve access to drugs, especially for emerging infectious diseases endemic to the continent45.
All of these agencies need to be able to stand alone.
In addition, there is an urgent need for global harmonization across regulatory authorities to address the substantial inequities in access to medicines.
Ideally, clinical trials for new therapies should be conducted globally, for access and generalizability46.
However, the reality is that clinical trials, including RCTs, cannot be conducted in every country to generate specific evidence for that country’s population.
Evidence generation using transportability analysis is gaining traction and refers to the ability to generalize inferences from a study sample in one country to a target population in another country where the study was not conducted47,48.
Transportability analyses may offer some evidence of external validity with implications for local regulatory and health technology assessments48.
Evidence generation in clinical research
Clinical studies of rare diseases
As an example, the FDA granted accelerated approval to alpelisib (Vijoice, Novartis) for adults and children over 2 years of age who require systemic therapy for PIK3CA-related overgrowth spectrum, which includes a group of rare disorders linked to mutations in the PIK3CA gene51.
Interestingly, efficacy was evaluated using a retrospective chart review of RWD from EPIK-P1 ( NCT04285723), a single-arm clinical study in which individuals with PIK3CA-related overgrowth spectrum received alpelisib as part of an expanded access program for compassionate use.
The application for this approval used the Real-Time Oncology Review pilot program52, which streamlined data submission before filing of the entire clinical application, and Assessment Aid53, a voluntary submission from the applicant to facilitate assessment by the FDA.
As a result, this application was granted priority review, breakthrough designation and orphan drug designation51.
As an example, the FDA granted accelerated approval to alpelisib (Vijoice, Novartis) for adults and children over 2 years of age who require systemic therapy for PIK3CA-related overgrowth spectrum, which includes a group of rare disorders linked to mutations in the PIK3CA gene51.
Interestingly, efficacy was evaluated using a retrospective chart review of RWD from EPIK-P1 (NCT04285723), a single-arm clinical study in which individuals with PIK3CA-related overgrowth spectrum received alpelisib as part of an expanded access program for compassionate use.
The application for this approval used the Real-Time Oncology Review pilot program52, which streamlined data submission before filing of the entire clinical application, and Assessment Aid53, a voluntary submission from the applicant to facilitate assessment by the FDA.
As a result, this application was granted priority review, breakthrough designation and orphan drug designation51.
N-of-1 trials
In the era of individualized genomic medicine, N-of-1 trials are emerging as a tool to study potentially fatal rare diseases.
The N-of-1 trial is a single-patient clinical trial using the individual person as a unit of investigation to evaluate the efficacy and/or adverse events of different interventions through objective data-driven criteria54.
For example, an antisense oligonucleotide therapy was designed for, and evaluated in, a single patient who had a fatal genetic neurodegenerative disorder known as CLN7 neuronal ceroid lipofuscinosis (a form of Batten’s disease)55.
Another patient (who happened to be a physician) with idiopathic Castleman’s disease refractory to IL-6-blocking therapy identified the causative molecular alteration in his own disease to develop a personalized therapy56.
In yet another example, rapid dose escalation with a selective RET inhibitor was evaluated in a single patient with highly refractory medullary thyroid carcinoma, to overcome a resistance mechanism specific to that patient57.
These sensational new drug discovery-translation paradigms raise important questions, such as what level of evidence is needed before exposing a human to a new drug, what evidence this approach might generate for the next patient and what challenges might exist with generalizability58.
The concept of medical analog patient-specific ‘digital twins’ is an emerging area of research that has the potential to combine polynomial data (mechanistic data, medical history, with the power of AI) and may perhaps serve to enhance N-of-1 trials in the future, to further personalize medicine37,59,60.
The concept of medical analog patient-specific ‘digital twins’ is an emerging area of research that has the potential to combine polynomial data (mechanistic data, medical history, with the power of AI) and may perhaps serve to enhance N-of-1 trials in the future, to further personalize medicine.
RWD and real-world evidence
One of the major criticisms of all clinical trial research is that clinical trials do not represent the ‘real-world’ population; often, the restrictive criteria of clinical trials and the limited analyses framed to answer specific questions may not apply to real-world patients.
A wide gap therefore exists between the trial world and the real world, and attempts have been made to close this gap61.
Conventional trials have been designed on the basis of the misconception that regulatory bodies may not accommodate more modern and diverse evidence from RWD, which is no longer the case61,62.
It is important to distinguish between RWD, which refers to data generated from routine, standard care of patients62, and real-world ‘evidence’ (RWE), which is the evidence generated from RWD regarding the potential use of a product.
RWE is generated by trial designs or analysis and is not restricted to randomized trials; instead, it comes from pragmatic trials and prospective and/or retrospective observational studies62,63.
In this purview of RWD and RWE, all stakeholders look to regulators for guidance.
Consequently, regulators have taken a hands-on approach and provided guidance and a comprehensive framework launched through the 21st Century Cures Act 62,64.
Moreover, the FDA uses RWD and RWE for postmarketing safety monitoring, and insurance agencies have started to use such data for coverage decisions62.
This has been necessitated by rapidly accelerating data input from multiple streams and layers into electronic health records, as well as wearables and biosensors, in parallel with new analytical capabilities (multimodal AI) to analyze the vast amount of data.
Moreover, the FDA uses RWD and RWE for postmarketing safety monitoring, and insurance agencies have started to use such data for coverage decisions62.
This has been necessitated by rapidly accelerating data input from multiple streams and layers into electronic health records, as well as wearables and biosensors, in parallel with new analytical capabilities (multimodal AI) to analyze the vast amount of data.
Evidence from synthetic or external control arms
RCTs are considered the gold standard for drug development and evidence as they allow for estimation of treatment effects that can be assigned to the experimental arm of interest.
The randomization in these studies curtails the concern for confounding bias by removing systematic imbalances between arms in measured and unmeasured prognostic factors65.
However, advances in the genomics of rare diseases and the discovery of rare oncogene-driven cancers have led to specific targeted therapies, for which evaluation in RCTs may not be feasible or ethical and may delay patient access to promising or lifesaving therapies.
In such cases, synthetic control arms are emerging as options for generating comparator arms that can ‘mimic’ the comparator arms of RCTs.
Synthetic control arms are external to the study in question, and most are derived from RWD65.
Moreover, RWD are obtained from electronic health records, administrative claims data, natural history registries and patient-generated data from many sources, including wearable devices65.
Synthetic control arms may also be generated from previous clinical trial data (single or pooled trials).
This is an emerging area primed for innovation as so much data are now available from multiple sources.
Synthetic control arms are external to the study in question, and most are derived from RWD.
Moreover, RWD are obtained from electronic health records, administrative claims data, natural history registries and patient-generated data from many sources, including wearable devices65.
Synthetic control arms may also be generated from previous clinical trial data (single or pooled trials).
NSCLC is increasingly being divided into small oncogene-driven subsets, making it more challenging to conduct randomized trials66, and recent developments in the NSCLC trial landscape illustrate the utility of synthetic control arms.
For instance, RET fusions are genomic drivers in 1–2% of NSCLCs, and pralsetinib is a selective RET-targeted therapy showing promising responses even in individuals with advanced disease.
The ARROW study ( NCT03037385) was a single-arm registrational trial, conducted globally, to evaluate pralsetinib in RET fusion-positive individuals with NSCLC67,68.
This trial showed a relative survival benefit with the drug when compared to an external standard-of-care control arm consisting of RWD cohorts derived from two Flatiron Health databases66.
A template for future studies of this nature using quantitative bias analyses showed that comparisons between the external control arm and the trial arm are robust and able to withstand issues such as data missingness, potentially poorer outcomes in RWD and residual confounding66.
Overall, the study provided evidence in favor of pralsetinib as a first-line treatment for RET fusion-positive NSCLC.
The use of synthetic control arms can accelerate drug development, and initial skepticism about them arose mainly from a lack of precedence and direction from regulatory authorities.
These concerns are now being dispelled as synthetic control arms have been used recently for drug approvals for ultra-rare diseases.
For example, neurofibromatosis is a rare disease seen in 1 in 3,000 births. Patients develop plexiform neurofibroma lesions that are painful and debilitating, causing motor and neuronal dysfunction.
The MEK inhibitor selumetinib was approved for pediatric patients with symptomatic, inoperable plexiform neurofibromas on the basis of a dataset of 50 patients from Selumetinib in Pediatric Neurofibroma Trial (SPRINT)-a single-arm phase 2 trial showing a durable objective response rate and improvements in functional symptoms65,69,70.
Comparator arms from two previously conducted trials provided evidence for the natural history of the disease and were submitted as an external control arm, which helped confirm that spontaneous regressions were uncommon and that the observed responses and symptom improvement represented a genuine treatment effect69.
Despite this progress, external control arms are still an emerging concept and they have mainly been used to investigate the natural history of disease and have not generally been included as primary evidence or in product labels.
However, in the future, I can envision such comparative effectiveness analysis and comparator arms as primary evidence to support drug approval.
Challenges mainly arise from data quality and data missingness, as well as uncertainty of whether external control data are fit for purpose.
However, some of these concerns can be mitigated by quantitative bias analysis and other methodologies66,71.
Pediatric clinical trials
Although pediatric research has been at the forefront of major advances in medicine (extracorporeal membrane oxygenation72is a notable example) and has pushed the boundaries of modern oncology (for instance, in treating pediatric leukemia), innovations in new drug development are often delayed.
Many rare and orphan diseases occur mainly in the pediatric population, and drug development in this population has always been operationally, ethically, statistically and methodologically challenging73,74.
This is compounded by the limited understanding of basic biology, the ontology of disease manifestations, and the acute and long-term safety of products73,74.
In addition, there is considerable off-label use of products in very young children, infants and neonates where clinical trials have not been feasible, and it is imperative that high-level evidence be generated by creative methods.
Programs such as the Best Pharmaceuticals for Children Act (in 2002) and the Pediatric Research Equity Act (in 2003), made permanent in 2012 under the FDA Safety and Innovation Act, have incentivized and enhanced the development of pediatric therapeutics73.
Innovative trial designs, RWD and leveraging data from other resources may help with risk-benefit assessment and drug approval, such as the approval for neurofibromatosis type 1 (NF1)73.
Many rare and orphan diseases occur mainly in the pediatric population, and drug development in this population has always been operationally, ethically, statistically and methodologically challenging.
Reimagining the future of clinical trials
- 1.AI
- 2.Partnerships in drug development
- 3.Social media and online community research
1.AI
The landscape of AI in medicine has transformed recently, and AI is poised to become ubiquitous.
Several RCTs have quantified the benefits of AI in specialties that use pattern recognition and interpretation of images, such as radiology (mammography and lung cancer screening), cardiology (interpreting electrocardiograms (EKGs), cardiac functional assessment and atrial fibrillation screening), gastroenterology (interpreting colonoscopies), pathology (cancer diagnosis), neurology (tracking disease evolution of amyotrophic lateral sclerosis and Parkinson’s disease), dermatology (diagnosing lesions) and ophthalmology (eye disease screening)75.
However, most AI research focuses on ‘clinical care delivery’ applications and not ‘clinical trial research’76.
However, most AI research focuses on ‘clinical care delivery’ applications and not ‘clinical trial research’.
The integration of AI into clinical trial research has been slower than expected, mainly owing to the (perceived) friction between AI versus human intelligence.
Nevertheless, trials of data generation and interpretation should be conducted, and AI should be used to augment human intelligence-not seen as something to replace it77.
Next-generation clinical trials using AI should consider AI + human rather than AI versus human scenarios75,78.
The clinical trial guidelines for protocols (Standard Protocol Items: Recommendations for Interventional Trials-Artificial Intelligence (SPIRIT-AI) extension) and publications (Consolidated Standards of Reporting Trials-Artificial Intelligence (CONSORT-AI) extension)79,80are intended to achieve standardized and transparent reporting for randomized clinical trials involving AI, and these are just the beginning of a new phase of clinical research modernization.
Given the time and cost involved in developing a drug, every failed drug in the market represents a considerable loss to the drug development ecosystem.
In addition, inferior trial designs, suboptimal patient recruitment, poor infrastructure to run trials, and inefficiency in trial conduct and monitoring have plagued the system for decades.
AI has the potential to augment all phases of drug development, from drug design to the complete drug development cycle (Fig. 1).
In addition, inferior trial designs, suboptimal patient recruitment, poor infrastructure to run trials, and inefficiency in trial conduct and monitoring have plagued the system for decades.
AI has the potential to augment all phases of drug development, from drug design to the complete drug development cycle
Clinical trial conduct is still rudimentary in many ways. For instance, in oncology trials, a few aspects of two-dimensional lesions are measured and followed over time and effectiveness of the drug is evaluated by shrinkage of these lesions.
Automated quantitative assessments and artificial neural networks can aid in automated rapid processing of multiple lesions81.
In cardiology trials, vital signs are measured once a week in clinic, and, in neurology, patient questionnaires are administered in clinic.
Now, these data can all be tracked dynamically in real time using wearable sensor technology.
The application of AI to such areas can have a transformational near-term impact.
In addition, pattern recognition using deep neural networks can help with reading scans, pathology images and EKGs, among others37,78.
The current evidence-based medicine pyramid represents the tip of the iceberg and barely provides shallow evidence to care for a generic patient (Fig. 4).
Hence, a deep synthesis and amalgamation of all available data is needed to achieve next-generation, ‘deep’ evidence-based medicine.
The main challenge in the next two decades will be to tap the potential of multidimensional evidence generation82 by extracting, collating and mining large sets of natural history data, genomics and all other omics analysis, all published clinical studies, RWD, data from ubiquitous smart devices and amassed data from the IoMT to provide next-generation evidence for deep medicine.
Hence, a deep synthesis and amalgamation of all available data is needed to achieve next-generation, ‘deep’ evidence-based medicine.
The main challenge in the next two decades will be to tap the potential of multidimensional evidence generation82 …
… by extracting, collating and mining large sets of natural history data, genomics and all other omics analysis, all published clinical studies, RWD, data from ubiquitous smart devices and amassed data from the IoMT …
… to provide next-generation evidence for deep medicine.
Fig. 4: Evidence-based deep medicine iceberg.
The current evidence-based medicine (EBM) pyramid represents the tip of the iceberg and barely provides enough shallow evidence to care for a generic patient. Hence, a deep synthesis and amalgamation of all available data is needed to achieve next-generation, deep evidence-based medicine. The main challenge ahead in the next two decades will be extracting, collating and mining large sets of natural history data, genomics and all omics analyses, all published clinical studies, RWD and amassed data from the IoMT to provide next-generation evidence for deep medicine. PRO, patient-reported outcomes.
2.Partnerships in drug development
Currently, the pharma industry is the main driver of drug development, and their expenditures far exceed investments from any national agency such as the National Institutes of Health61.
There are two domains of clinical trials.
The first of these is from ‘big pharma’, which uses CROs to run trials; such trials are very often approved for registration by the FDA.
The second domain encompasses academic clinical trials, which often operate on a very limited budget, do not often evaluate new compounds and, thus, rarely result in FDA registration.
In this era of reduced federal funding for research, more partnerships are needed for drug development.
Academic centers and community sites are crucial for patient enrollment; however, a siloed mentality has impacted drug development and delayed access to lifesaving therapies.
Therefore, collaborations among specific disease organizations, academic institutions, federal agencies and patient advocacy groups are crucial for betterment of the health of populations (Fig. 3).
Because the pharma industry is hesitant to invest huge amounts with limited financial return, especially in rare diseases, federal agencies have developed programs to incentivize rare disease drug development1.
Moreover, disease-focused organizations have collaborated with the pharma industry, federal agencies and academia to form ‘venture philanthropy’ with risk-sharing financial models to de-risk drug development1.
Many academic institutions are entering into risk-sharing strategic alliances with the pharma industry to collaborate across preclinical and clinical development phases.
Such successful innovative partnership models have set a precedent in diseases such as cystic fibrosis, multiple myeloma, type 1 diabetes mellitus and other rare diseases1.
These collaborations have effectively catalyzed innovation through all phases of drug development and provided a compelling reason to sustain and foster more of these sorts of programs.
3.Social media and online community research
Social media outlets (Twitter, Facebook and so on) can influence patient accrual in clinical trials.
They can strongly influence and address historical clinical trial challenges, including the lack of awareness among patients and physicians about available trials and the lack of community engagement.
More than 4.48 billion people use social media globally, and this number is projected to increase to almost 6 billion in 2027 (ref. 83).
Over 70% of Americans are on social media, including rural dwellers and adolescent and young adult populations who have always been under-represented in clinical trials.
Although many older adults do not use social media, their caregivers are likely to.
People with terminal diseases often self-experiment with drugs, and online patient communities can provide environments for sharing and monitoring such drug usage.
This can allow for observational studies to be planned around quantitative, internet-based outcome data.
For example, researchers developed an algorithm to dissect the data reported on the PatientsLikeMe website by people with amyotrophic lateral sclerosis who experimented with lithium carbonate treatment84.
This analysis reached the same conclusion as an ensuing RCT, suggesting that data from online patient behavior can help accelerate drug development and evaluate the effectiveness of drugs already in use.
An increase in engagement from patients and patient advocacy groups can aid patient education and outreach and can facilitate patient-partnered research, …
… as well as allowing for incorporation of patients’ perspectives in the design of clinical research-ultimately generating research that is driven by the needs of real people with the disease under investigation.
Moreover, social media breaks open silos dividing researchers and clinicians, creating enormous potential to influence all areas of medicine85.
Conclusion
The success of future clinical trials requires a fundamental transformation in how trials are designed, conducted, monitored, adapted, reported and regulated to generate the best evidence.
The status quo model is unsustainable.
Instead, preventive, personalized, pragmatic and patient-participatory medicine is needed, and paradigm shifts are required to get there via sustainable growth.
Silos need to be broken.
Standards of care and clinical trials are currently viewed in different realms; however, the overarching goal of both is to improve health outcomes.
The COVID-19 pandemic created an opportunity to observe how routine clinical care and clinical trials can work synergistically to generate evidence86. Pragmatic platform trials such as the RECOVERY trial should be a model and guide for trial efficiency and real-time impact.
Current paradigms must be continuously challenged by emerging technology and by all stakeholders (the new generations of scientists, physicians, the pharma industry, regulatory authorities and, most importantly, patients).
Disruptive innovation should lead to every clinical site being a research site, with all necessary quality checks and research as part of the standard of care.
The healthcare system should be integrated into an intuitive RWE-generation system, with clinical research and clinical care going hand in hand.
Beyond an ad hoc creative flash of genius (necessitated by a pandemic), sustained momentum will be needed to leverage the knowledge gained from programs such as ‘Operation Warp Speed’ (initiated by the US government to accelerate COVID-19 vaccine development).
My personal view is that every major disease needs a ‘Moonshot’ program and every rare disease should have an ‘Operation Warp Speed’-both with clearly identified, sustainable goals to improve population health and address equity, diversity and global access to therapies.
Methodological advances and future AI-based analyses of all data will provide deep evidence to realize the goal of personalized medicine- that is, to offer the right treatment to the right patient at the right time.
References & additional information
See the original publication
About the author(s) & affiliation(s)
Vivek Subbiah
Department of Investigational Cancer Therapeutics, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Division of Pediatrics, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
MD Anderson Cancer Network, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Acknowledgements
V.S. is an Andrew Sabin Family Foundation fellow at the University of Texas MD Anderson Cancer Center. V.S. acknowledges the support of the Jacquelyn A. Brady Fund. V.S. thanks the team at Draw Impacts for figures.
V.S. is supported by the US National Institutes of Health (NIH) (grants R01CA242845 and R01CA273168); the MD Anderson Cancer Center Department of Investigational Cancer Therapeutics is supported by the Cancer Prevention and Research Institute of Texas (grant RP1100584), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy (grant 1U01CA180964), NCATS (Center for Clinical and Translational Sciences) (grant UL1TR000371) and the MD Anderson Cancer Center Support (grant P30CA016672).
Cite this article
Subbiah, V. The next generation of evidence-based medicine. Nat Med (2023). https://doi.org/10.1038/s41591-022-02160-z
Originally published at https://www.nature.com on January 16, 2023.
RELATED PUBLICATIONS







