the health strategist
institute for strategic health transformation
& digital technology
Joaquim Cardoso MSc.
Chief Research and Strategy Officer (CRSO),
Chief Editor and Senior Advisor
October 9, 2023
What is the message?
The article discusses a breakthrough in drug discovery through the use of machine learning and high-performance computing.
Researchers from the University of Eastern Finland, in collaboration with industry partners and supercomputers, conducted one of the largest virtual drug screening campaigns.
The traditional approach to screening large compound libraries for potential drug molecules was time-consuming and required significant computational resources.
The study demonstrated that machine learning, specifically a tool called HASTEN, can significantly accelerate the screening process by predicting docking scores for compounds more efficiently than traditional methods.
This collaboration between academia and industry, along with the power of supercomputing, led to promising results and the release of valuable datasets to benefit future drug discovery efforts.

Executive Summary
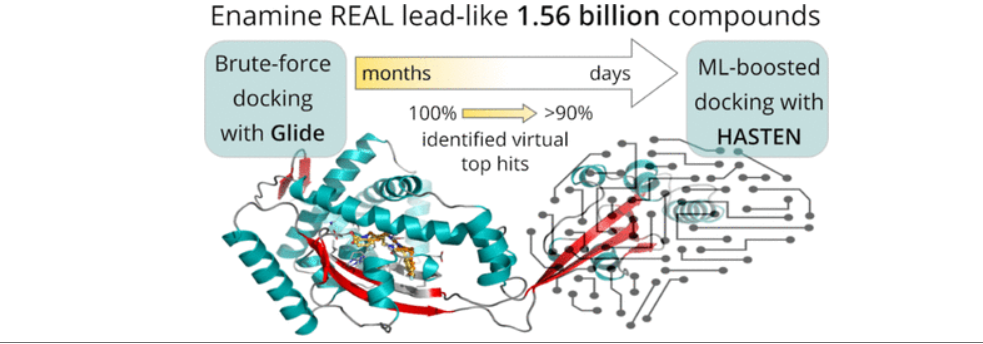
Boosting virtual screening with machine learning allowed for a 10-fold time reduction in the processing of 1.56 billion drug-like molecules. Researchers from the University of Eastern Finland teamed up with industry and supercomputers to carry out one of the world’s largest virtual drug screens.
In their efforts to find novel drug molecules, researchers often rely on fast computer-aided screening of large compound libraries to identify agents that can block a drug target. Such a target can, for instance, be an enzyme that enables a bacterium to withstand antibiotics or a virus to infect its host. The size of these collections of small organic molecules has seen a massive surge over the past years. With libraries growing faster than the speed of the computers needed to process them, the screening of a modern billion-scale compound library against only a single drug target can take several months or years – even when using state-of-the-art supercomputers. Therefore, quite evidently, faster approaches are desperately needed.
In a recent study published in the Journal of Chemical Information and Modeling, Dr Ina Pöhner and colleagues from the University of Eastern Finland’s School of Pharmacy teamed up with the host organisation of Finland’s powerful supercomputers, CSC – IT Center for Science Ltd. – and industrial collaborators from Orion Pharma to study the prospect of machine learning in the speed-up of giga-scale virtual screens.
Before applying artificial intelligence to accelerate the screening, the researchers first established a baseline: In a virtual screening campaign of unprecedented size, 1.56 billion drug-like molecules were evaluated against two pharmacologically relevant targets over almost six months with the help of the supercomputers Mahti and Puhti, and molecular docking. Docking is a computational technique that fits the small molecules into a binding region of the target and computes a “docking score” to express how well they fit. This way, docking scores were first determined for all 1.56 billion molecules.
Next, the results were compared to a machine learning-boosted screen using HASTEN, a tool developed by Dr TuomoKalliokoski from Orion Pharma, a co-author of the study. “HASTEN uses machine learning to learn the properties of molecules and how those properties affect how well the compounds score. When presented with enough examples drawn from conventional docking, the machine learning model can predict docking scores for other compounds in the library much faster than the brute-force docking approach,” Kalliokoski explains.
Indeed, with only 1% of the whole library docked and used as training data, the tool correctly identified 90% of the best-scoring compounds within less than ten days.
The study represented the first rigorous comparison of a machine learning-boosted docking tool with a conventional docking baseline on the giga-scale. “We found the machine learning-boosted tool to reliably and repeatedly reproduce the majority of the top-scoring compounds identified by conventional docking in a significantly shortened time frame,” Pöhner says.
“This project is an excellent example of collaboration between academia and industry, and how CSC can offer one of the best computational resources in the world. By combining our ideas, resources and technology, it was possible to reach our ambitious goals,” continues Professor Antti Poso, who leads the computational drug discovery group within the University of Eastern Finland’s DrugTech Research Community.
Studies on a comparable scale remain elusive in most settings. Thus, the authors released large datasets generated as part of the study into the public domain: Their ready-to-use screening library for docking that enables others to speed up their respective screening efforts, and their entire 1.56 billion compound-docking results for two targets as benchmarking data. This data will encourage the future development of tools to save time and resources and will ultimately advance the field of computational drug discovery.
DEEP DIVE
Machine Learning-Boosted Docking Enables the Efficient Structure-Based Virtual Screening of Giga-Scale Enumerated Chemical Libraries
ACS Publications
Toni Sivula, Laxman Yetukuri, Tuomo Kalliokoski, Heikki Käsnänen, Antti Poso, and Ina Pöhner
September 1, 2023
Abstract

The emergence of ultra-large screening libraries, filled to the brim with billions of readily available compounds, poses a growing challenge for docking-based virtual screening. Machine learning (ML)-boosted strategies like the tool HASTEN combine rapid ML prediction with the brute-force docking of small fractions of such libraries to increase screening throughput and take on giga-scale libraries. In our case study of an anti-bacterial chaperone and an anti-viral kinase, we first generated a brute-force docking baseline for 1.56 billion compounds in the Enamine REAL lead-like library with the fast Glide high-throughput virtual screening protocol. With HASTEN, we observed robust recall of 90% of the true 1000 top-scoring virtual hits in both targets when docking only 1% of the entire library. This reduction of the required docking experiments by 99% significantly shortens the screening time. In the kinase target, the employment of a hydrogen bonding constraint resulted in a major proportion of unsuccessful docking attempts and hampered ML predictions. We demonstrate the optimization potential in the treatment of failed compounds when performing ML-boosted screening and benchmark and showcase HASTEN as a fast and robust tool in a growing arsenal of approaches to unlock the chemical space covered by giga-scale screening libraries for everyday drug discovery campaigns.
Introduction
Virtual screening (VS) approaches utilizing molecular docking are a common choice in the early stages of structure-based drug discovery projects. Typically, their objective is to find the initial small molecule hits predicted to bind to a previously unexplored target, or to discover novel scaffolds in an unbiased way when confirmed binders of the studied target are already known. Especially in the initial screening steps, VS often utilizes large and diverse screening libraries to pool the most promising candidates from a large chemical space. (1−3) Chemical libraries of off-the-shelf or readily synthesizable (make-on-demand) compounds are popular to ensure that the docking predictions can be validated in biochemical assays in a timely manner without the need to factor in potentially time-consuming organic synthesis. In recent years, such libraries have continuously grown and today often cover a vast chemical space with compound numbers on the billion scale. (4) For example, at the beginning of this project, the Enamine REAL library of lead-like compounds had a size of 1.56 billion (March 2021), whereas the current version has 3.93 billion compounds (August 2023). (5) What is a huge leap forward in terms of access to diverse chemical space and straightforward validation of docking-based hypotheses, at the same time becomes a challenge for docking-based VS: since conventional brute-force docking visits every compound in a chemical library, screening billions of compounds is often no longer feasible, both in terms of the required time and the computational power.
On the other hand, structure-based VS of such libraries has produced hits of exceptional quality for several targets (see, e.g., studies by Lyu et al., (2) Stein et al., (3) and Kaplan et al. (6)). One key study to motivate the work with such large libraries was presented by Lyu et al. (2) Their docking of around 100 million compounds to two targets, β-lactamase AmpC, and D4 dopamine receptor, did not only result in the discovery of novel ligands for both targets, but they made critical observations for many studies to follow: first, they found that if docking enriches true positives over decoys on the small scale, enrichment can be expected to be translated to the ultra-large scale. Second, they demonstrated that, at least for the D4 receptor, the novel ligands could not have been found by, for example, docking only cluster representatives instead of the full library. Consequently, as compound libraries grow even bigger, there is a clear need for faster and more efficient methods to screen entire ultra-large libraries.
There are various approaches to tackle the computational expense associated with billion-scale structure-based VS. One strategy is to grow compounds from fragments instead of docking full-size molecules and thus avoiding the enumeration of large numbers of compounds. (7,8) Alternatively, several strategies that recently gained traction for boosting docking-based screening rely on iterative approaches utilizing machine learning (ML). (9−12) The idea is simple, yet powerful: a small fraction of a large chemical library is docked by conventional means and used as training data for a ML model. The model then either classifies the remainder of the library in “virtual hits” and “non-hits” (DeepDocking (9)) or aims to predict the docking scores for the entire library (e.g., MolPAL, (10) Glide Active Learning, (11) and HASTEN (12)). This way, when not every compound needs to be docked, ML-boosted screening approaches can handle ultra-large libraries in a fraction of the time, providing the opportunity to explore the vast chemical landscape of giga-scale libraries.
ML acceleration with the help of regression models has, for instance, been previously achieved by a random forest (RF) model (option in MolPAL), (10) an ensemble of RF and a Graph Convolutional Neural Network (Glide Active Learning), (11) a simple feed-forward neural network (MolPAL), (10) and by the message-passing neural network Chemprop (MolPAL (10) and HASTEN (12)).
In this work, we rely on the tool HASTEN (macHine leArning booSTEd dockiNg), which uses Chemprop to predict docking scores in an iterative approach: (12) in brief, HASTEN starts from an initial random compound selection from a large chemical library for a first conventional docking run and trains a ML model with the obtained data. Then, scores for the full compound library are predicted and used to rank all compounds. The best-ranked compounds next get selected for docking in the following iteration. This deliberate bias toward top-scoring compounds has been shown to result in excellent recall for data sets on the million scale. (12) We aimed to investigate the applicability and performance of the HASTEN approach on even larger data sets, where the training data already reaches the million scale and predictions on the giga-scale cover billions of compounds.
To benchmark the performance of the ML-boosted approach on giga-scale chemical libraries, such libraries first need to be screened using standard brute-force docking to obtain the baseline data. Such docking data on the giga-scale has so far at least rarely been made publicly available: to date, the only example we are aware of are the Covid-19 screening results obtained with AutoDock-GPU on the Oak Ridge National Laboratory Summit computer. (13,14)
Herein, we used the fast Glide high-throughput virtual screening (HTVS) method in one of the largest conventional docking campaigns performed to date, to provide a brute-force docking baseline for analysis and comparison with our ML-boosted approach. (15) We selected two distinct targets based on ongoing academic drug discovery projects.
The first target, the SurA protein, is a periplasmic chaperone found in Gram-negative bacteria. SurA has prolyl-peptidyl isomerase activity and is involved in the transport and maturation of several outer membrane proteins. (16−19) Loss of SurA activity has been shown to render resistant bacterial strains sensitive to antibiotics, making SurA an interesting target in combating antibiotic resistance. (18,20)
Our second target was the cyclin G-associated kinase (GAK): a serine/threonine kinase, serving as a regulator of clathrin-mediated endocytosis and clathrin trafficking. (21−23) GAK represents an important host factor involved in the regulation of viral entry and assembly of different RNA viruses, such as hepatitis C, dengue, and Ebola virus, and is of interest as an anti-viral target. (24−26)
Our study aims to benchmark and demonstrate the potential of ML-boosted screening with HASTEN for giga-scale applications, showcasing the speed-up, robustness, and successful recall of the majority of top-scoring compounds compared to the brute-force docking results for our two targets. We further demonstrate how large numbers of compounds that fail to dock successfully can hamper the HASTEN approach and discuss different options to handle such “failed” compounds, ultimately resulting in a novel screening protocol for HASTEN.
Finally, to support future screening approaches, we release a prepared version of the Enamine REAL lead-like screening library used in this study in a Glide-compatible format. Additionally, we release our full giga-scale docking results as benchmarking data sets for the future development and improvement of ML-boosted screening procedures.
Approach
Computational Infrastructure
Computational resources were provided by CSC─IT Center for Science Ltd. (27) All calculations were performed on the CSC supercomputers Mahti (Atos BullSequana XH2000) and Puhti (Atos BullSequana X400), both running Red Hat Enterprise Linux Server release 7.9.
Ligand preparation and conventional docking steps were carried out with Mahti. Mahti features 1404 CPU nodes, each equipped with two AMD Rome 7H12 CPUs with 64 physical cores capable of two hardware threads running at 2.6 GHz base frequency and 256 GBs of system memory and Lustre parallel storage system providing a peak file I/O performance of 1.5 GB/s.
ML was conducted on CSC Puhti, which is equipped with 80 GPU nodes, in which each has four Nvidia Volta V100 GPUs. Puhti GPU nodes further feature two Intel Xeon Cascade Lake 20-core CPUs running at 2.1 GHz, 384 GB of system memory, and a local 3.6 TB NVMe disk.
Screening Database Preparation
The Enamine REAL lead-like (ERLL) library, containing a total of 1.56 billion compounds (March 2021), was selected for the giga-scale screening. (5) All included compounds have lead-like properties, with molecular weight ≤460 Da, Slog P −4.0 to 4.2, number of hydrogen bond acceptors <10 and donors <5, number of ring systems ≤4, and rotatable bonds ≤10. The library was obtained in the ChemAxon extended SMILES format and converted to regular SMILES using RDKit v2021.03.5, retaining the stereochemical information of the compounds where applicable. (28,29)
To reduce the variation in docking times between different subsets, compound order first was randomized, and SMILES were then evenly divided into 20 subsets. Ligand 3D structure preparation for docking was carried out with Schrödinger LigPrep (Schrödinger Suite 2021-1). (30) Up to eight tautomers per compound and four stereoisomers per tautomer for a target pH of 7.0 ± 1.5 were generated, and compound geometries were energy-minimized with the OPLS_2005 forcefield.
Pre-prepared compounds were next collected into Schrödinger Phase databases for use during docking and future studies. (30−32) Coordinates were stored in the compact internal coordinate representation, and a single conformation was generated with rapid sampling from each input compound during the phase revise step. To enable parallel processing within the CSC system wall time limits, the 20 input files were further split into a total of 781 individual phase databases of approx. 4.8 million compounds each.
Receptor Preparation and SiteMap Analysis
Structures of SurA (PDB-ID 1M5Y, chain A) (33) and GAK (PDB-ID 4Y8D, chain A) (34) were retrieved from the RCSB Protein Data Bank. (35) The Schrödinger Protein Preparation Wizard was used for structure preparation, hydrogen addition, and bond order assignment. Missing side chains were added with Prime. (30) Crystallographic agents and water molecules were deleted. State generation for the original crystallographic ligand in the GAK structure was performed with Epik for pH 7.0 ± 2.0. Amino acid protonation states for pH 7.0 were assigned with PROPKA, and the hydrogen bonding network was optimized. Receptors were then subjected to a restrained energy minimization in the OPLS_2005 forcefield until the heavy atom rmsd compared to the previous minimization step fell below 0.3 Å.
For a comparison of the pocket properties of the two chosen targets, binding pocket properties were computed with Schrödinger SiteMap. (30,36,37) The prepared GAK structure was directly subjected to SiteMap analysis, using only the site defined by the crystallographic ligand. SiteMap was run with default parameters (at least 15 site points per site, a more restrictive definition of hydrophobicity, standard grid, and cropping at 4 Å from the nearest site point).
The SurA apo-structure was first subjected to a 1 μs molecular dynamics simulation with Desmond and frames from the last 200 ns were analyzed with SiteMap to identify probable and druggable small molecule-binding sites (calculating up to five top-ranked sites per run; data not shown). The selected site was located in the crevice between N- and C-terminal core and P1 domains of SurA (for further discussion of the SurA domain architecture and pockets, see Calabrese et al. (38)).
Receptor Grid Generation and Docking
Grid generation for SurA used the frame with the most druggable and consistently identified site, as described above (kindly provided by T. Kronenberger), and the receptor grid with a size of 30 Å3 was centered on the site centroid. For GAK, the grid center was defined as the centroid of the crystallographic ligand. Both grids were prepared with the OPLS_2005 forcefield. For GAK, additionally, a hydrogen-bonding constraint on the hinge-region amide (Cys126 backbone amide hydrogen) was set up.
Conventional docking of the 1.56 billion Enamine REAL lead-like library was carried out with Schrödinger Glide v9.0 in the HTVS mode. (15,30) Van der Waals radii of nonpolar ligand atoms were scaled to 0.8 with a charge-cutoff of 0.15 e (default), and nonplanar amide conformations were penalized in both targets. Additionally, for GAK, the hydrogen-bonding constraint on the hinge-region amide of Cys126 was used. With the HTVS mode, the OPLS_2005 forcefield was used, and a single pose per ligand was collected after subjecting five poses to post-docking minimization.
Simulated ML-Boosted Docking with HASTEN
The ML-boosted docking was simulated using the simu-dock mode in a local implementation of HASTEN v0.2 (optimized for CSC Puhti). (12) For ML, Chemprop v1.3.1 (with Python 3.8.12) inside a singularity-container was used. (39) Briefly, simu-dock allows the use of pre-generated docking data instead of actual docking in the HASTEN procedure. Whenever a compound is selected for docking by the algorithm, the pre-generated results of the conventional docking study will be loaded. Since scores are only added to the training data when compounds were selected for docking, the system exhibits identical behavior to screening with HASTEN when run including the brute-force docking steps.
In the first iteration, training was initialized with a random selection of 0.1% of the full library (1.56 million compounds). The selected compound subset was split randomly into training, validation, and test sets amounting to 80, 10, and 10% of the selected compounds, respectively. When no docking score was obtained during the conventional docking run (i.e., the compound did not dock successfully or failed to satisfy the constraint in GAK), an arbitrary failed score of +5.0 or 0.0 was applied, or failed compounds were excluded entirely from the training data. We performed one round of HASTEN with each treatment of failed compounds for each target. For SurA, experiments with a failed score of +5.0, and for GAK, experiments with the exclusion of failed compounds, were repeated in triplicate.
We used default parameters for regression in Chemprop to predict the docking score, except for the batch_size parameter, which was increased to 250 (from default: 50) to speed up training (see Supporting Information for a list of parameters). Once the training was completed, the scores for all 1.56 billion compounds in the library were predicted from their SMILES strings. Compounds were then ordered by the predicted score, and docking scores of the top-ranked 0.1% of compounds, that were not previously selected for docking, were loaded to simulate their conventional docking. All loaded docking scores were used to train the ML model from scratch during the next iteration, and the procedure was repeated nine times, which corresponds to docking 1% of the 1.56 billion input library by conventional means (compare also the schematic workflow in Figure 1).

Additionally, a HASTEN run with excluded failed compounds and a smaller docking fraction of only 0.01% per iteration was carried out for each target (all other settings were kept the same). This way, 10 iterations corresponded to docking only 0.1% of the 1.56 billion compounds. For GAK, the run with the reduced docking fraction was extended to a total of 25 iterations.
Analysis of the Results
Recall values of the top 100, 1000, and 10,000 compounds were computed after every iteration. We define recall herein as the fraction of the true top-scoring 100, 1000, and 10,000 compounds when ranked by brute-force docking results, that were found using the ML model and selected for docking by HASTEN up until the current iteration.
Chemical similarities between the virtual hits and compounds in their training data set were evaluated using Tanimoto similarities calculated from 2048 bit Morgan fingerprints. Fingerprints were created using chemfp and RDKit, and Tanimoto similarities were calculated with chemfp. (29,40)
To assess the consistency of results obtained in repeated predictions, three replicates with a different random selection of compounds for the initial training set were used. The overlap of the compounds selected for docking by each model was calculated by counting which compounds among the top-scoring 100, 1000, and 10,000 compounds were selected by one, two, or three of the replicates after each iteration.
Results and Discussion
Chosen Targets SurA and GAK have Distinct Binding Pocket Properties
One objective in selecting targets for our case study was to ensure their distinct binding pocket properties. Since docking and, in particular, scoring success is target-dependent, any ML approach trained on docking scores will inherently reflect the same target dependence. (41) In line with that, previous work with HASTEN, utilizing 12 literature targets, has already revealed different performances of the same protocol against different targets. (12)
However, having two targets with distinct properties assessed on the giga-scale would, for the first time, indicate whether the ML-boosted HASTEN performs equally well for both targets when benchmarked against the brute-force docking backdrop on the giga-scale and thus highlight whether potential additional dependencies arise from the choice of the ML method when employed on HASTEN’s intended use-scale. To assess the pocket properties, we first computed pocket descriptors with Schrödinger SiteMap, as summarized in Table 1.
Table 1. SurA and GAK Binding Pocket Properties Computed with SiteMap: DScore, Druggability Score (Values of 1.0 or Greater are Generally Considered Druggable); (37) Hydrophobic, Hydrophilic, Hydrophobic, and Hydrophilic Character of the Site, Respectively (a Value of 1.0 Represents the Average for Tight-Binding Sites); (36) don/acc, the Ratio of Hydrogen Bond Donors to Hydrogen Bond Acceptors
| target | DScore | volume [Å3] | hydrophobic | hydrophilic | don/acc |
|---|---|---|---|---|---|
| SurA | 1.15 | 363 | 1.52 | 0.80 | 1.25 |
| GAK | 1.03 | 507 | 0.66 | 1.02 | 0.91 |
The selected binding pocket of SurA was smaller than the GAK pocket (363 vs 507 Å3, Table 1) and displayed a high hydrophobicity (hydrophobic 1.52, Table 1). Furthermore, the binding site features more hydrogen bond donors than acceptors (donor acceptor ratio 1.25, Table 1). The GAK target was, on the other hand, more hydrophilic (hydrophilic 1.02, Table 1) and had a higher proportion of acceptors in the binding pocket (donor acceptor ratio 0.91, Table 1).
In conclusion, the two targets chosen for our case study display diverging binding pocket properties and can consequently be expected to favorably interact with different chemical scaffolds, thereby allowing us to investigate the method’s performance on the giga-scale in two distinct screening scenarios.
Ready-to-Use Glide-Compatible Screening Library and Giga-Scale Brute-Force Docking to SurA and GAK
This is an excerpt. For more information, read the original publication.
ML-Boosted Giga-Scale Screening of SurA and GAK
For accelerating the screening process with the help of ML, we used the tool HASTEN, which has been previously validated with FRED and Glide SP docking results on the million scale. (12)
HASTEN aims to identify the top-scoring compounds rather than attempting a generalized prediction of docking scores for a given target. By iteratively selecting compounds with the best predicted scores, the training data will be progressively enriched in both true positives (already ranked correctly by the model) and false positives (ranked highly by the model, although the compounds dock poorly). This will improve HASTEN’s capability of identifying true top-scoring compounds with every iteration. Figure 1 summarizes our adapted procedure for the giga-scale screening.
We first started from an initial random selection of 0.1% of the 1.56 billion compounds. Previous work with HASTEN for million-scale data typically involved the selection of 1% of the library on each iteration, but since our training data at 0.1% already exceeds one million compounds, we decided to instead aim for a final total docking of 1% of the giga-scale library.
The brute-force docking step was simulated by utilizing the pre-generated docking data for the complete screening library: docking scores of compounds that were selected for docking by the algorithm were loaded directly. Only scoring data of selected compounds was considered, which ensures HASTEN to run as if the brute-force docking step had been performed as part of the workflow. Docking scores and corresponding compound SMILES were used as input data for training a ML model with Chemprop. (39) Next, with the generated model, docking scores for the full ERLL library were predicted, and compounds were ranked by their predicted scores. During the next iteration, docking results for the top-ranked 0.1% of the compounds were added to the training data. This process was repeated nine times to end with a total training data set amounting to 1% of the full giga-scale data set.
Reducing the required number of compounds to dock to 1% of the full library lowered the total time spent on ligand preparation and SurA docking to around 1 day and 4 h when utilizing 640 CPUs of the CSC Mahti supercomputer and 20 h for the GAK target (including ligand preparation; 17,628 and 12,668 CPU hours for SurA and GAK, respectively).
The ML steps of the HASTEN protocol (Figure 1, blue boxes) consumed an additional 203–335 h for ML model training and prediction. Training took 143 h on a single Nvidia Volta V100 GPU of CSC Puhti. Prediction steps were distributed over 10 GPUs, resulting in a total prediction time of 52–184 h, depending on whether multiple predictions were run in parallel on one GPU or each GPU ran only a single instance of Chemprop. While a single instance per GPU was overall fastest, Chemprop, in our environment, utilized only a fraction of the available GPU compute power and memory, with its main bottleneck being compound featurization on the CPU. Thus, running four instances per GPU enabled higher throughput with the same hardware and constituted a better utilization of the computing resources on the supercomputer.
Removing the necessity of docking 99% of the entire giga-scale library allows the HASTEN procedure to complete the screening in around 10–14 days for each of the two targets. Additionally, depending on available resources, this approach enables a user to balance the computational load associated with a giga-scale screening project between CPUs (most docking tools) and the faster GPUs (for ML).
Adjusting the Failed Score or Excluding Failed Compounds from the Training Data can Improve the Recall
When docking was unable to produce a docking score, for example because all sampled poses were energetically unfavorable, the original HASTEN protocol associated an arbitrary positive docking score with the affected SMILES strings to mimic a positive and therefore unfavorable energy. We started out with such a “failed score” of +5.0 for the SurA target and observed excellent recall values.
Herein, we define recall as the number of true 100, 1000, and 10,000 top-scoring compounds according to the brute-force docking approach, that had also been selected for docking by HASTEN. With SurA and a failed score of +5.0, we were able to recall around 95, 90, and 85% of the top-scoring 100, 1000, and 10,000 compounds, respectively (see Figure 2 for top 1000 and Table S1). Using the same approach with the GAK target, on the other hand, resulted in recalls of only 70, 67, and 59% of the top-scoring 100, 1000, and 10,000 compounds, respectively (Figure 2 and Table S2).

One major difference between the data sets is the number of failed compounds: while for SurA less than 3% of all compounds in the ERLL library fails to dock (total failed: 42,149,150 compounds), for GAK, 45% are not docked successfully (704,564,272 compounds, compare also the overall distribution of docking scores in Figure S1). Consequently, a total of 46,846 failed compounds were selected into the HASTEN training data for SurA (final training data size >15 million compounds, of which 0.3% had the failed score). In contrast, for GAK, 1,471,958 failed compounds were part of the training data, that is around 9% of the total training data had the failed score of +5.0.
A reason for the high number of failing compounds may lie in the treatment of hydrogen-bonding constraints in Glide: with the active hinge region amide constraint, any compound with no hydrogen bond accepting group will be directly excluded from evaluation and receive the failed score. For any other compound, the initial placement will depend on its hydrogen bond acceptors: with constraint fulfillment being the first objective, rather than optimizing compound orientations for enclosure in the pocket, partially exposed compound poses can occur since they fulfill the constraint, albeit being energetically unfavorable and thus, likewise, receiving the failed score. The constraint thus forces compounds that would receive a favorable score when docked without the constraint to be evaluated in a different, more unfavorable receptor context.
We hypothesize that the large fraction of failed compounds in the training data drives the learning process in the ML step toward primarily identifying failed compounds to minimize the chosen metric (RMSE) rather than picking up smaller differences between the successfully docked compounds. Importantly, since failed compounds share features with those that dock successfully and because failure to dock can be an artifact of the docking methodology, the model can only imperfectly predict which compounds will fail. As a consequence, when their failed score is far outside the distribution of scores from successful dockings (see Figure S1), this will drive the model into associating higher, less favorable scores with features observed in failed compounds.
To improve the recall for the GAK data set, we next attempted to adjust the failed score parameter: motivated by our hypothesis, we set the failed score to 0.0 (closer to other obtained docking scores) to reduce the emphasis on failed compounds during training, which improved the recall of the top-scoring 100, 1000, and 10,000 compounds by 9–13% when compared to the failed score of +5.0 (compare Figure 2 and Table S2). We tested the same approach for SurA, which did, however, not consistently improve the recall (Table S1).
Finally, we assessed the complete removal of all failed compounds from the training data. Notably, the exclusion of all failed compounds resulted in a recall of 94, 90, and 84% of the top 100, 1000, and 10,000 true virtual hits for GAK, which is similar to the results achieved initially with SurA (compare Figure 2 and Tables S1 and S2). Moreover, using the same approach for SurA also improved the recall, albeit only slightly by about 1–2% (Table S1).
Comparing the number of failed compounds selected for docking during each iteration between the initial GAK screen with a failed score of +5.0 and the screen where failed compounds were excluded supports our hypothesis that the model learns to recognize compounds that will fail: the initial random selection of both runs includes around 708,000 failed compounds. In later iterations, only about 10% of this initial number get selected when failed compounds were considered with a failed score of +5.0 (see Figure S2). On the other hand, when excluding failed compounds completely and thus not providing the model with any training reference to recognize features of failed compounds, around 30–40% of the initial number of failed compounds get added during each iteration (see Figure S2).
When lowering the failed score or excluding failed compounds, the improvement of recall suggests that the learning process is instead driven by the identification of good-scoring compounds to minimize the RMSE metric. Furthermore, the validation and test set RMSE values per iteration suggest that a failed score closer to the mean or dropping of the failed compounds in both cases overall improves the model (see Figure S3).
In summary, we showed that in certain docking scenarios, the complete removal of failed compounds from the training data appears to improve the model quality. Depending on the case, it can also greatly enhance the recall (GAK) or show only a minor impact on the recalled compounds (SurA). In particular, dropping failed compounds is likely beneficial when a large proportion of evaluated compounds fail to dock successfully. This protocol modification can also add to the speed-up of model training as there is less data to process. Our case study identified the treatment of failed compounds or their assigned score as factors that can improve recall in ML-boosted screening campaigns with HASTEN/Chemprop.
Prospects of Further Speed-up by Hyperparameter Choice and Protocol Modifications
This is an excerpt. For more information, read the original publication.
HASTEN Robustly Identifies the Same Compounds Irrespective of the Initial Random Selection
As a final step in our giga-scale assessment, we aimed to verify that HASTEN robustly recalls the majority of the top-scoring compounds irrespective of the initial random compound selection. To study the overlap of recalled compounds, we performed our ML-boosted screening experiment with a docking fraction of 0.1% per iteration in triplicate, with each run starting from a different random set of compounds.
For SurA, we repeated the run with the original protocol and a failed score of +5.0, and for GAK, the run with failed compounds excluded from the training data. As can be seen in Figure 3, some variation occurs during the first iterations, especially for the top 100 and top 1000 virtual hits. However, recalls converged in the later iterations. The final recall of the top scoring 100, 1000, and 10,000 SurA virtual hits was on average 94, 90, and 85%, respectively. For GAK, average recalls were 94, 90, and 84% (see Tables S7 and S8). Thus, in conclusion, all three runs for both targets had a highly similar recall.

We further investigated the overlap in compound selection: as shown in Figure 4 for the top 1000 virtual hits, the different HASTEN runs initially have no overlap, and from iteration 2 rapidly converge into largely the same final selection of compounds. Of the 90% recalled top 1000 virtual hits, around 30% were selected by all three replicates already on iteration 2 and around 87–88% were recalled consistently during the HASTEN runs by all three replicates (see Figure 4, black bars and Venn diagrams for iterations 2–10 in Figures S9 and S10 in the Supporting Information). Thus, our case study indicates that a single run of HASTEN is sufficient, and no major recall benefit could be gained from repeating experiments with a different initial random selection, at least in a setting with training data on the million scale. Additionally, the swift convergence into the same selection of compounds indicates that the robustness of the HASTEN approach can still be assumed when stopping on an earlier iteration.

Conclusions
Our case study of the two targets SurA and GAK confirmed the applicability of the ML-boosted docking tool HASTEN on the so far unprecedented giga-scale. Our comparison with the corresponding brute-force docking results demonstrated comparable recall for the two distinct targets, identifying, for example, 90% of the true top-scoring 1000 virtual hits, although a modified protocol was necessary in the case of GAK. Given its benefit for the recall in both targets, this novel, modified protocol, which drops failed compounds from the training data, has now been made the default in HASTEN as of version 1.1.
Herein, our primary objective was the investigation and benchmarking of HASTEN for the speed-up of screening efforts on the giga-scale. While all tools that utilize ML for faster VS were ultimately developed to be able to process modern giga-scale screening libraries, to the best of our knowledge, so far no tool has been benchmarked against a billion-scale docking ground truth. Our work thus makes HASTEN the first application that underwent a benchmarking effort on the intended use-scale.
With the growth trend of chemical libraries in mind, novel approaches to facilitate giga-scale screening will likely continue to be developed. Benchmarking such approaches on ultra-large data sets remains challenging since the generation of giga-scale reference data is prohibited by time and resource consumption in many settings. We thus also release our full giga-scale docking results to provide others, seeking to predict docking scores from SMILES, with aptly sized benchmarking data sets.
Our “ground truth” and reference in this work were brute-force docking results generated with the particularly fast Glide HTVS protocol. Increased docking speed is typically achieved by limiting conformational sampling and is thus associated with a scoring accuracy trade-off and “false negatives”. The more robust a scoring protocol is in terms of fewer “mislabeled” compounds, the easier it should be for the ML model to associate a previously unseen compound structure with an appropriate score. In our hands, using the smaller 0.01% docking fraction of HTVS results did not achieve the same recall as with the larger docking fraction of 0.1%. However, as also backed by previous work with HASTEN and a small-scale comparison of Glide SP and HTVS results (data not shown), we conclude that equally powerful models to the ones achieved in this work could be generated with smaller training data stemming from more robust docking approaches, such as Glide SP. Importantly, this allows the approach to be tailored to the available resources: a user can either generate more training data with a less robust, but faster, docking methodology or rely on fewer docking results for training when utilizing more robust and computationally expensive methods.
One should keep in mind that ligand preparation and explicit brute-force docking steps remain the most time-consuming part of the procedure and should be kept to the required minimum. To support and potentially speed up future screening efforts involving brute-force docking campaigns, we release the entire prepared ERLL library as Glide-compatible, randomized ready-to-use screening databases.
Our results indicated additional time-saving potential in strategies such as earlier stopping, which we found often associated with only a minor drop in the recall of virtual hits. Depending on the available resources, as well as the desired outcome of the screening campaign, we herein outlined possible modifications to the HASTEN protocol for maximum speed-up or maximum recall of top-scoring hits.
It is important to note that we herein compare against brute-force docking as our baseline. Docking scores have known limitations in their ability to rank compounds correctly and identify true actives. When an approach can enrich actives over property-matched decoys on a small scale, this has been previously shown to translate to the ultra-large scale. (2) For the GAK target, where experimentally confirmed actives were known, we validated the enrichment ability of the Glide HTVS protocol. It should be noted that the HASTEN protocol is, by definition, most useful when the chosen docking approach can, as in the case of GAK, successfully enrich in true actives.
In our case study, an excellent recall was achieved when using the default parameters of the ML engine Chemprop. It is however possible that both speed and recall could be further improved by performing hyperparameter optimization. While Chemprop provided excellent results in our case study, we acknowledge that it may not always be the ML engine of choice for every screening scenario.
In conclusion, HASTEN represents a robust approach to identify the bulk of the top-scoring virtual hits of a brute-force giga-scale docking campaign in a reduced time frame by reducing the required docking calculations by 99% (or more). In this work, HASTEN has been benchmarked on the giga-scale and shown to represent a viable strategy for ML-boosted docking to join a growing arsenal of methods designed to tackle the challenges associated with screening giga-scale libraries in everyday drug discovery.
References
This is an excerpt. For more information, read the original publication.
Authors and Affiliations
Corresponding Author
Ina Pöhner – School of Pharmacy, University of Eastern Finland, Kuopio FI-70211, Finland;
Authors
Toni Sivula – School of Pharmacy, University of Eastern Finland, Kuopio FI-70211, Finland
Laxman Yetukuri – CSC─IT Center for Science Ltd., Espoo FI-02101, Finland
Tuomo Kalliokoski – Computational Medicine Design, Orion Pharma, Orionintie 1A, Espoo FI-02101, Finland
Heikki Käsnänen – Computational Medicine Design, Orion Pharma, Orionintie 1A, Espoo FI-02101, Finland
Antti Poso – School of Pharmacy, University of Eastern Finland, Kuopio FI-70211, Finland; Department of Pharmaceutical and Medicinal Chemistry, Institute of Pharmaceutical Sciences, Eberhard Karls University, Tübingen DE-72076, Germany; Cluster of Excellence iFIT (EXC 2180) “Image-Guided and Functionally Instructed Tumor Therapies”, University of Tübingen, Tübingen DE-72076, Germany; Tübingen Center for Academic Drug Discovery & Development (TüCAD2), Tübingen DE-72076, Germany
Originally published at https://pubs.acs.org